
Ling-Ya Peng, Guang-Ning Pan, Wen-Kai Chen, Xiang-Yang Liu*, Wei-Hai Fang, Ganglong Cui*. Angew. Chem. Int. Ed., 2024, 63, e202315300.
The efficient conversion of excess CO2 into various high-added value chemicals can solve the greenhouse effect caused by excessive CO2 emissions and alleviate the energy crisis. Even though CO2 can be efficiently converted into organic substances through photosynthesis in nature, the artificial capture and conversion of CO2 is still very challenging due to its stability and kinetic inertness, which has attracted much research interest. In the present work we have employed the highly accurate multi-reference CASPT2 method to comprehensively explore the excited-state properties and photophysical processes of a PNNP-type Ir catalyst, namely [Ir(III)H]+; while, its subsequent reduction reactions to either HCOOH or CO are systematically studied using the efficient DFT method. The corresponding rates of radiative, nonradiative, and electron transfer processes are also calculated.
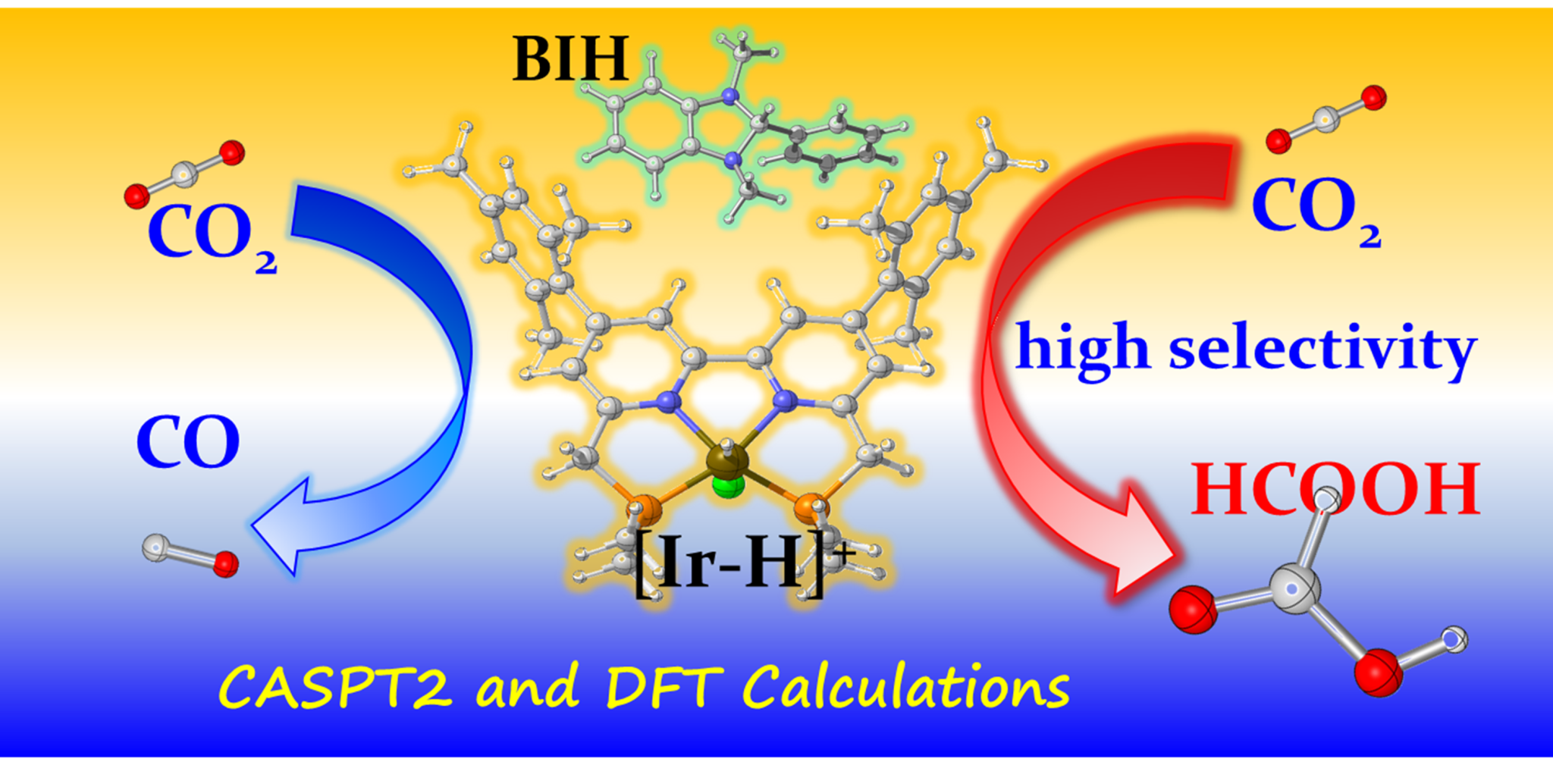
Figure 1. Photocatalytic reduction of CO2 by the Ir(III) complex [Ir-H]+.
The photophysics of the [Ir(III)H]+ can be well explained by a four-state model including S0, 1MLCT, 3LE, and 3MLCT. The precursor 3MLCT state can be populated from the initially excited 1MLCT state via both the efficient direct and 3LE-mediated intersystem crossings. Starting from this 3MLCT state, the CO2 reduction reactions diverge. In the presence of BIH, an electron sacrificial agent, an excited-state single electron transfer (SET) to the 3MLCT state of [Ir(III)H]+ generates a one-electron-reduced species (OERS), which initiates a series of reduction reactions to HCOOH. In comparison, a cascade of reduction reaction to CO starts from a deprotonated Ir(I) species generated from [Ir(III)H]+ in the 3MLCT state. In the photocatalytic cycle, either to HCOOH or CO, nonadiabatic transitions are frequently seen and play an essentially nonnegligible role. We also found that the high selectivity to HCOOH should originate from two aspects, a relatively smaller free-energy barrier in the rate-limiting step and the pretty much efficient SET leading to OERS. More interestingly, in the reduction reaction to either HCOOH or CO, BIH•+ is found to effectively reduce energy barriers of several important reactions. Methodologically, the present work shows that highly accurate multi-reference electronic structure methods are indispensable for studying certain stages of photocatalytic reactions of organometallic compounds involving electronically excited states, multiple spin states, open-shell intermediates, and remarkable nonadiabatic effects. Finally, the gained mechanistic insights should help interested chemists to understand, regulate, and design photocatalytic CO2 reduction reaction of similar function-integrated molecular photocatalyst.
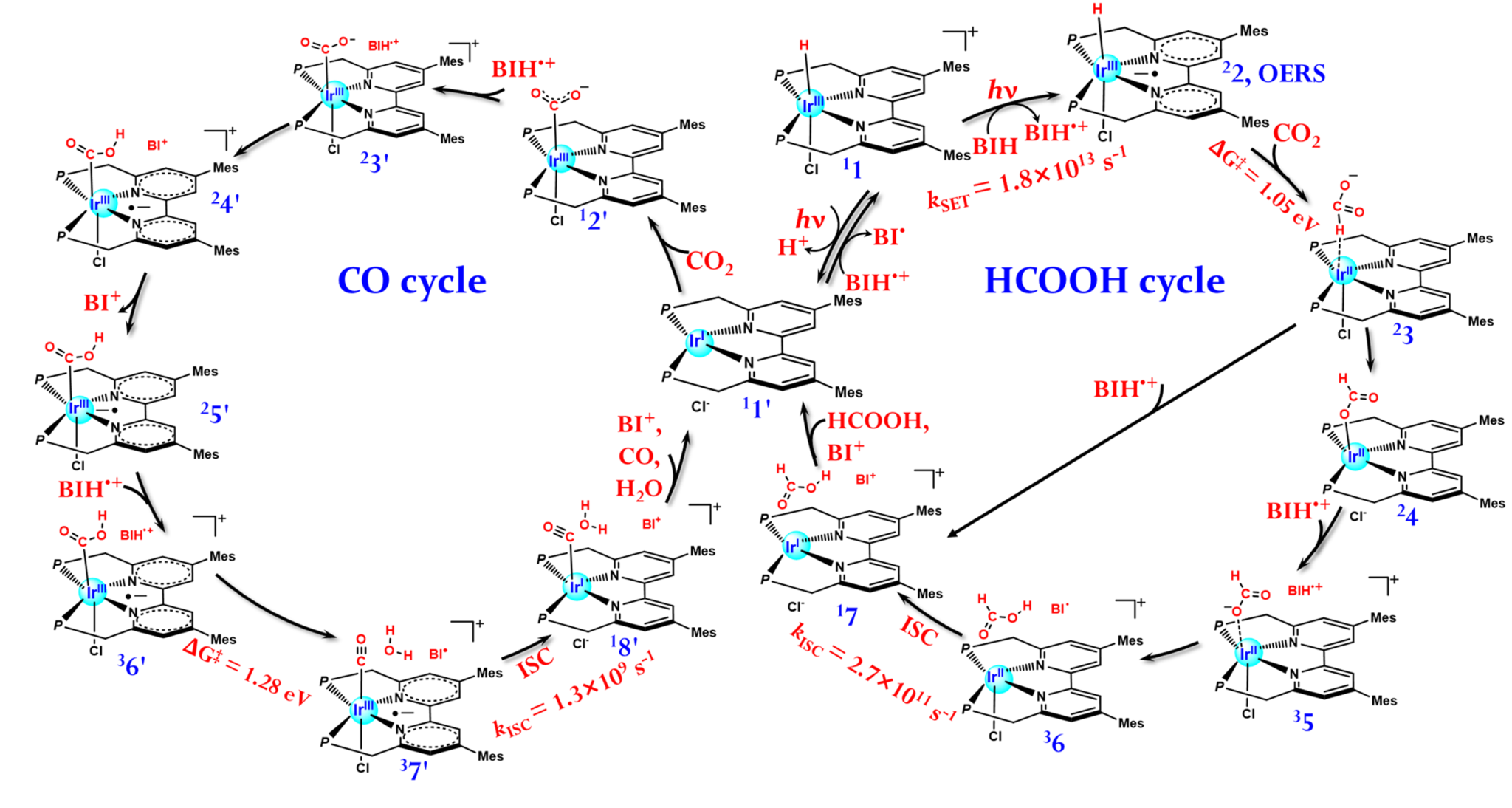
Figure 2. Proposed photocatalytic reduction reaction mechanism for the HCOOH and CO formation.
First Author: Peng Lingya
Correspondence Authors: Prof. Cui Ganglong, Beijing Normal University, Assoc. Rsr. Liu Xiangyang, Sichuan Normal University
Full Text Link: https://doi.org/10.1002/anie.202315300





 Latest Updates
Latest Updates

